
EUDAMED improves transparency and coordination of information on medical devices. However, it often excels in its complexity. The main component of this complexity is its six modules, of which only three are released as of today. There is no release date scheduled for the remaining three modules yet. However, EU information materials are often difficult to understand and sometimes raise more questions than they answer.
Nevertheless, a general understanding of EUDAMED is essential. After all, its use will become mandatory after a transitional phase, once the entire system is fully functional. Insufficient preparation and non-compliance with the regulations can thus lead to lost sales if products are not allowed to be released onto the market. However, it should be mentioned that not every module is relevant for all users. Therefore, this article presents the basics of the six EUDAMED modules and explains their functions.
Actor Registration
Although it has already been activated, actor registration is not yet mandatory. However, they must have registered by the date of EUDAMED activation. Economic operators include manufacturers, authorized representatives, system & procedure pack producers, and importers. Furthermore, all users of a company, who will work in EUDAMED, must register. It should be noted that a company registers where it is located and not where the products are marketed. After registration, an SRN (Single Registration Number) is created and serves as a unique identification of the economic operator. Furthermore, they must have at least one person with the required medical device expertise in their organization. This person is then responsible for compliance with the MDR regulations. The aim of the “Actor registration” module is to create the greatest possible transparency for customers of medical devices. The manufacturers of these devices can be identified via this EUDAMED module.
UDI/Devices Registration
The purpose of this EUDAMED module is the identification of medical devices by unique serial numbers, the Unique Device Identification, or short: UDI. Here manufacturers enter the most important information of their products, that are for sale in the European Union, into EUDAMED. These are to be made available to the public. The aim of “UDI/Devices registration” is to ensure the traceability of individual batches, the inspection of goods in quality management, and identification in logistics. Registration is carried out in accordance with the European Medical Devices Nomenclature (EMDN). A distinction is made here between “Regulation Devices”, “Legacy Devices” and “Other Devices”.
Notified Bodies and Certificates
A Notified Body is an organization for testing the requirements of medical devices before they are placed on the market. They are authorized to do so by an EU member state. Notified bodies are for example TÜV Rheinland or DEKRA. They are also called conformity assessment bodies and issue the necessary certificates to allow manufacturers to sell their products. The Notified Bodies are obliged to register the issued certificates, as well as related changes and additions in EUDAMED. All information and certificates are also available to the public here.
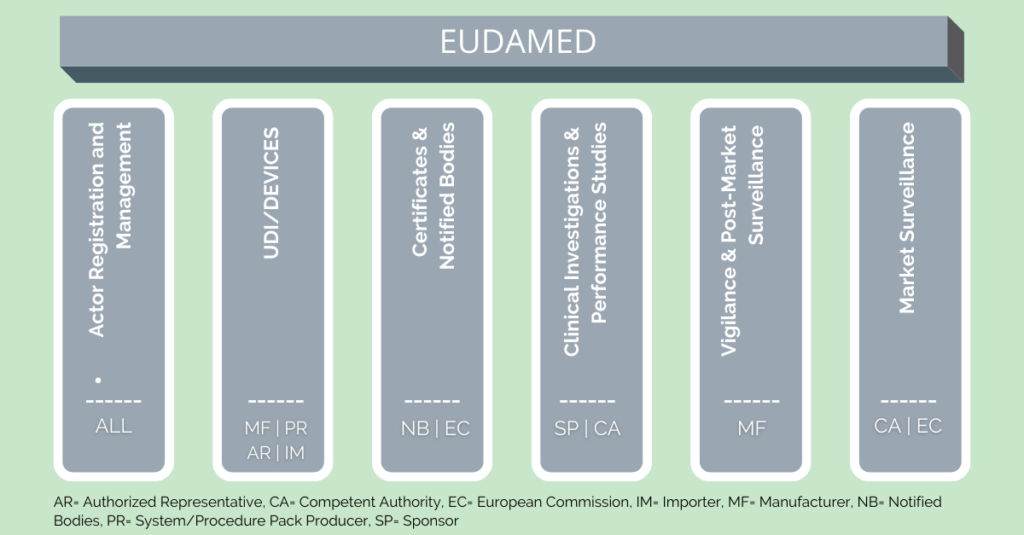
Clinical Investigations and Performance Studies
In alignment with EUDAMED, the products registered under it are partly subjected to clinical evaluations. This entails a systematic examination of whether the products provide what they promise and whether they are safe for patients. Tests of this kind are necessary if too little clinical data is available on the products. In this way, sufficient data are collected to enable the products to be sold on the European market.
Vigilance and Post-Market Surveillance
The EUDAMED post-market surveillance module (short: PMS) is self-explanatory. It is part of the EUDAMED vigilance system. By vigilance, the Medical Device Regulation (MDR) refers to the monitoring and reporting system of medical devices. Since manufacturers want to bring a product onto the market that is as safe as possible, these products need to undergo many tests. These are monitored by competent authorities and notified bodies. However, some risks only emerge after a certain period. The goal of PMS is to systematically detect these risks as the products are used, monitor their performance in use, and detect overlooked risks. In addition, the risk-benefit assessment takes place consistently, and recalls can be organized quickly in case of doubt.
Market Surveillance
As a part of market surveillance, the responsible authorities regularly check the functionality and safety of medical devices that are already on the market. Document reviews, physical inspections, and laboratory tests are conducted for this purpose. Counterfeit and unsafe products are thus removed from the market to ensure the safety of their users.
The EUDAMED modules are more complex than this article illustrates. It is only meant to serve as an introduction on the way to compliance. A partner with years of experience like tracekey can help to achieve compliance with the modules for the different actors. In addition, the European Commission announced an extension of the transition period for EUDAMED in October 2023.